Suggested protocol for nuclei isolation
Below is a suggested protocol for nuclei isolation. Many other protocols are available that might have already been optimized for the type of cells/tissue you are working with; please have a look at publications or protocols published online as well.
Buffers
| Components | Final concentration | Volume for 1ml |
|---|---|---|
| Tris-HCl (1M, pH 7.4) | 10 mM | 10 µl |
| NaCl (5 M) | 10 mM | 2 µl |
| MgCl2 (1 M) | 3 mM | 3 µl |
| BSA (10 %) | 1% | 100 µl |
| Tween 20 (10 %) | 0.1% | 10 µl |
| Nuclease-free water | - | 875 µl |
| Components | Final concentration | Volume for 1ml |
|---|---|---|
| Wash buffer | 980 µl | |
| Nonidet P40 Substitute (10%) | 0.1% | 10 µl |
| Digitonin (1%) | 0.01% | 10 µl |
Transposition mix
| Components | Volume per reaction |
|---|---|
| 2x Tagment DNA Buffer (TDB) | 2.5 |
| Tagment DNA Enzyme 1 (TDE) | 0.5 |
| Nuclease-free water | 2 |
| Total | 5 |
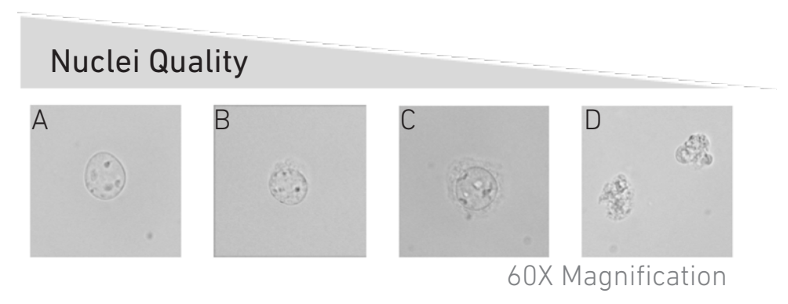
Nuclei QC
The quality of nuclei should be assessed with a microscope. The figure below (taken from 10x Userguide: CG000365_DemonstratedProtocol_NucleiIsolation_ATAC_GEX_Sequencing_RevB) illustrates nuclei with different integrities.
NUCLEI ISOLATION
Isolate single cell suspension (at least 100.000 cells) with an appropriate protocol, or start with ~3-5mg of (frozen) tissue.
When working with cells:
If you FACsort the cells, pre-load 5 µl of cell medium or PBS into the tube.
Centrifuge cells at 300 rcf for 5 min at 4°C. [choose the speed appropriate for your cell type]
Remove supernatant. ~5 µl may remain.
Add 100 µl chilled Lysis Buffer (Table 2) and gently pipette mix three times.
Without further incubation, centrifuge directly at 500 rcf for 5-10 min2 at 4°C.
When working with (frozen) tissue:
Put tissue on tube with lysis buffer, on ice. If tissue is frozen, let it thaw before grinding.
Gently grind the tissue with appropriate pestle, until no large clumps are visible. Avoid over-grinding to not break apart nuclei. A good starting point is between 4-8 strokes.
Without further incubation, centrifuge directly at 500 rcf for 5-10 min2 at 4°C.
Remove supernatant without disrupting the nuclei pellet.
Add 45 µl chilled WB (Table 1) to the tube and gently pipette the mix three times. Optionally, simply flick the tube a few times; the nuclei should resuspend easily after 3-4 taps.
Quality control step: Check nuclei quality under microscope. Mix 8 µl trypan blue (0.4% solution) with 2 µl of the nuclei suspension3,4 and transfer the mix to a Cell Counting Chamber for counting nuclei and checking nuclei quality (under a 40x objective, at least). Nuclei should be round with intact nuclear membrane, no aggregates and no debris.
Centrifuge the remaining nuclei suspension at 500 rcf for 5 min at 4°C.
(Meanwhile, prepare transposition mix)Remove most of the supernatant with a P100/200 pipette, then remove the remaining liquid with a 10µl pipette without touching the bottom of the tube, to avoid dislodging the nuclei pellet.
Resuspend the nuclei pellet in 5 µl transposition mix. Incubate reaction at 37°C for 30 minutes in a thermocycler (lid heated to 50°C).
Purify the tagmented DNA (e.g. Qiagen MinElute Reaction Cleanup Kit) and store the sample at -20°C.
It is advised to test different detergent concentrations. For this, lysis buffer can be diluted with wash buffer to 1:5, 1:10, 1:20 or 1:50 for example. (1:10 dilution is a good starting point)
Do not reduce spinning time to less than 5 minutes.
You can mix 5 µl trypan blue + 5 µl nuclei mix if you have sufficient starting material or only want to check nuclei quality. The more nuclei you can observe, the better you can estimate the quality. Alternatively, you can add a DNA-intercalating dye such as propidium iodide to check for nuclei integrity and free floating DNA.